ROCS-like shape overlap in rdkit
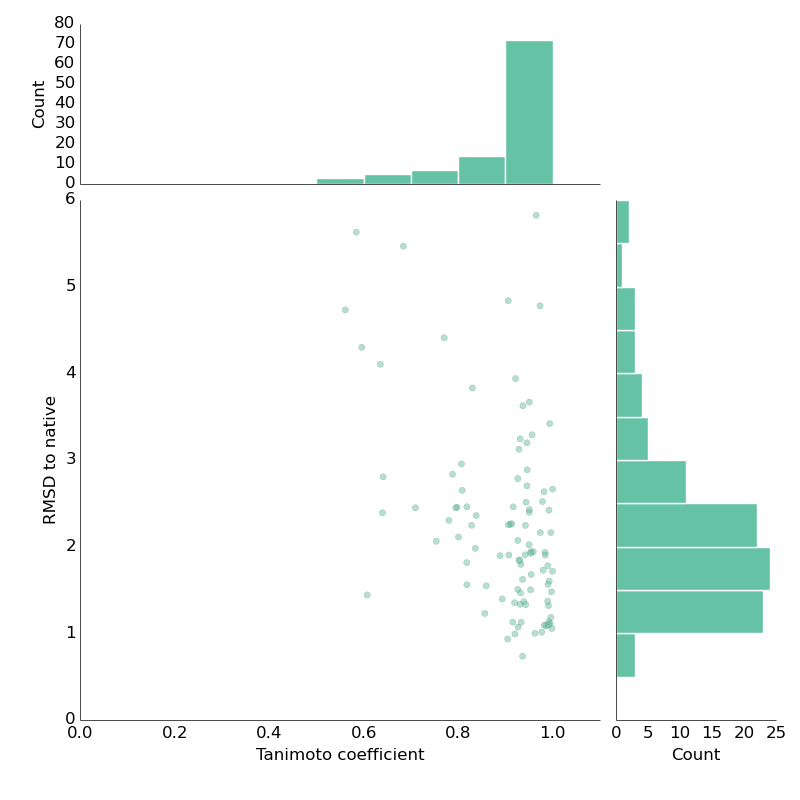
One year ago, I gave a brief talk at the RDKit user group meeting in Cambridge. As usual, I ranted about how it would be fantastic to have robust open-source computational chemistry tools. In particular, a commonly used tool from the 80s – gaussian-based shape overlap for pairs of molecules – was only covered by a single open-source package. So, together with help from some friends (big shout out to Greg Landrum, rdkit) I set out to change that. Took almost a year but we're getting there! The only open-source package doing shape overlap was shape-it from silicos-it. We basically ripped the code out of it and incorporated it into rdkit. The aim was to provide python-scriptable shape-overlap tool. The silicos-it is run by Hans de Winter – big thanks to him – I'm not sure what's going on now but the silicos-it webpage down. Greg did most (or certainly a _lot_) of work cleaning up the code. Now, we're finally moving towards testing against standard benc
