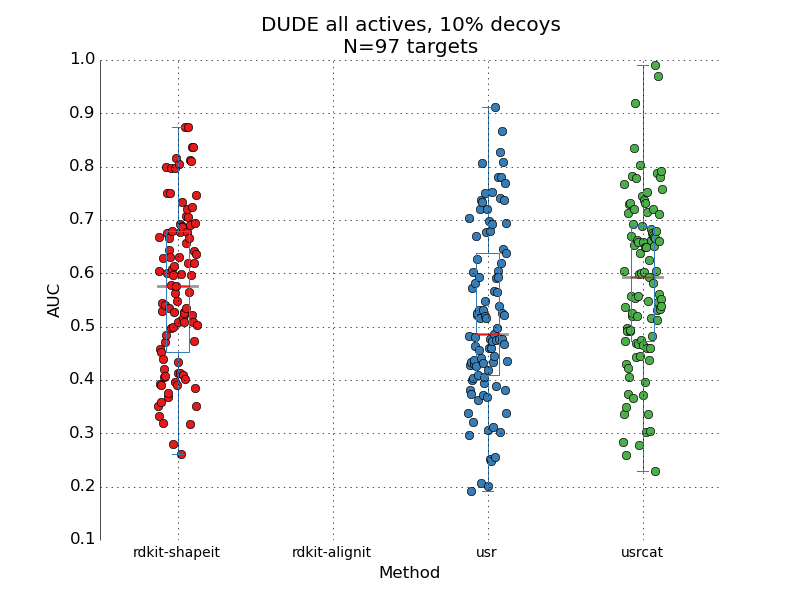
Performance of a popular docking code

This post is a quick note on a performance of a commercial docking code, as measured across the entire DUDE dataset. For around a 100 protein targets, the code is supposed to rank active compounds higher than decoys compounds – separate poppy seeds from sand if you like. Let me start by saying that I'm pretty impressed with what I've seen. Starting this side-project, I assumed that any docking code can be expected to have an AUC of around 0.6-0.7 measured on a standard benchmarking set (such as DUDE). I think that's largely true of free codes such as Autodock vina or rdock. But here we're looking at a piece of code from a major commercial vendor and it performs beyond my expectation. I'm not disclosing the name of the code, since there may have been something in the academic license that prevents such benchmarking studies from being published (a "gag order" effectively, in case somebody is measuring the code "incorrectly"). AUC is estima